28 KiB
Machine Learning para corrección de errores en datos de secuenciación de ADN
- Introducción
- Estado del arte
- Objetivos
- Métodos
- Resultados
- Conclusiones
- Futuras mejoras
- Bibliografía
Introducción
El ácido desoxirribonucleico (ADN) y el ácido ribonucleico (ARN) son los repositorios moleculares de la información genética. La estructura de cada proteína, y en última instancia de cada biomolécula y componente celular, es producto de la información programada en la secuencia de nucleótidos de una célula. La capacidad de almacenar y transmitir la información genética de una generación a otra es una condición fundamental para la vida. Un segmento de una molécula de ADN que contiene la información necesaria para la síntesis de un producto biológico funcional, ya sea una proteína o un ARN, se denomina gen. El almacenamiento y la transmisión de información biológica son las únicas funciones conocidas del ADN cite📖lehninger.
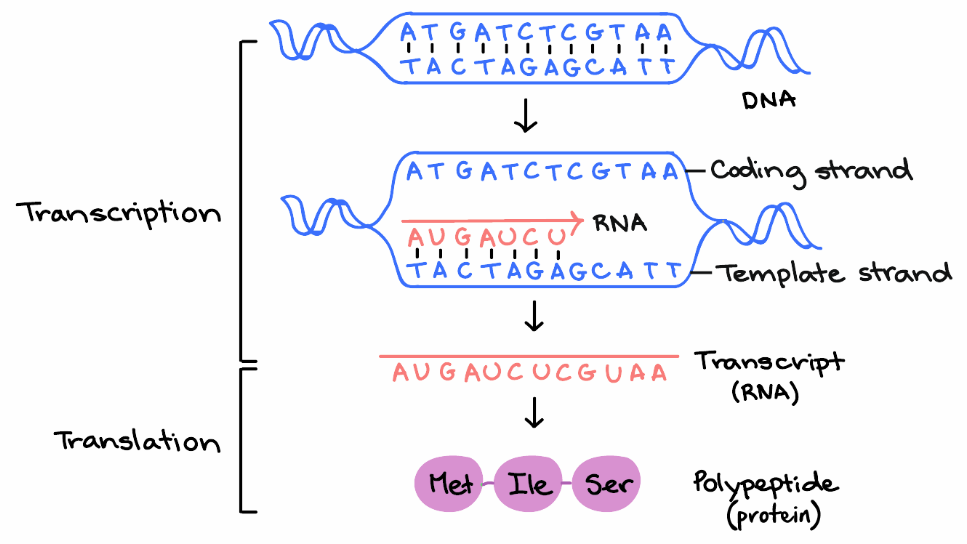
Tanto el ADN como el ARN son ácidos nucleicos, formados por cadenas de nucleótidos. Los nucleótidos son las unidades monoméricas de los ácidos nucleicos. El ADN contiene las bases adenina (A), guanina (G) citosina (C) y timina (T), mientras que el ARN contiene A, G y C, pero tiene uracilo (U) en lugar de timina (T) cite📖211898.
Hay muy pocos principios firmes en biología. A menudo se dice, de una forma u otra, que la única regla real es que no hay reglas, es decir, que se pueden encontrar excepciones a cada principio fundamental si se busca lo suficiente. El principio conocido como el Dogma central de la biología molecular parece ser una excepción a esta regla de excepción ubicua cite:CRICK1970. El dogma central de la biología molecular establece que una vez que la información ha pasado a proteína no puede volver a salir; \ie la transferencia de información de ácido nucleico a ácido nucleico, o de ácido nucleico a proteína puede ser posible, pero la transferencia de proteína a proteína, o de proteína a ácido nucleico es imposible cite:crick1958protein.
Las proteínas se producen mediante el proceso de traducción, que tiene lugar en los ribosomas y está dirigido por el ARN mensajero (ARNm). El mensaje genético codificado en el ADN se transcribe primero en ARNm, y la secuencia de nucleótidos del ARNm se traduce en la secuencia de aminoácidos de la proteína. El ARNm que especifica la secuencia de aminoácidos de la proteína se lee en codones, que son conjuntos de tres nucleótidos que especifican aminoácidos individuales cite📖211898. El código genético se muestra a continuación:
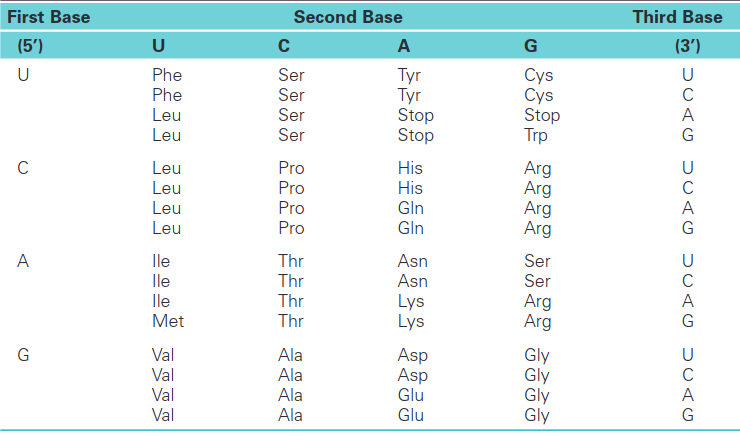
Por lo tanto, si elucidamos la información contenida en el ADN, obtenemos información sobre las biomoléculas que realizan las diferentes tareas fisiológicas y metabólica (e.g. ARN, proteínas).
Secuenciación de ADN
La secuenciación de ADN es el proceso mediante el cual se determina el orden de los nucleótidos en una secuencia de ADN. En los años 70, Sanger ηl desarrollaron métodos para secuenciar el ADN mediante técnicas de terminación de cadena cite:Sanger5463. Este avance revolucionó la biología, proporcionando las herramientas necesarias para descifrar genes, y posteriormente, genomas completos. La demanda creciente de un mayor rendimiento llevó a la automatización y paralelización de las tareas de secuenciación. Gracias a estos avances, la técnica de Sanger permitió determinar la primera secuencia del genoma humano en 2004 (Proyecto Genoma Humano). Cabe destacar que se estima que la secuencia final cubría aproximadamente el 92% del genoma cite:InternationalHumanGenomeSequencingConsortium2004.
Sin embargo, el Proyecto Genoma Humano requerió una gran cantidad de tiempo y recursos, y era evidente que se necesitaban tecnologías más rápidas, de mayor rendimiento y más baratas. Por esta razón, en el mismo año (2004) el National Human Genome Research Institute (NHGRI) puso en marcha un programa de financiación con el objetivo de reducir el coste de la secuenciación del genoma humano a 1000 dólares en diez años cite:Schloss2008. Esto estimuló el desarrollo y la comercialización de las tecnologías de secuenciación de alto rendimiento o Next-Generation Sequencing (NGS), en contraposición con el método automatizado de Sanger, que se considera una tecnología de primera generación.
Técnicas de secuenciación de alto rendimiento
Estos nuevos métodos de secuenciación proporcionan tres mejoras importantes: en primer lugar, en lugar de requerir la clonación bacteriana de los fragmentos de ADN, se basan en la preparación de bibliotecas de moléculas en un sistema sin células. En segundo lugar, en lugar de cientos, se producen en paralelo de miles a muchos millones de reacciones de secuenciación. Finalmente, estos resultados de secuenciación se detectan directamente sin necesidad de electroforesis cite:vanDijk2014.
Actualmente, se encuentran en desarrollo las tecnologías de tercera generación de secuenciación (Third-Generation Sequencing). Existe un debate considerable sobre la diferencia entre la segunda y tercera generación de secuenciación, la secuenciación en tiempo real y la divergencia simple con respecto a las tecnologías anteriores deberían ser las características definitorias de la tercera generación. Aquí consideramos que las tecnologías de tercera generación son aquellas capaces de secuenciar moléculas individuales, negando el requisito de amplificación del ADN que comparten todas las tecnologías anteriores cite:HEATHER20161.
Estas nuevas técnicas han demostrado su valor, con avances que han permitido secuenciar el genoma humano completo, incluyendo las secuencias repetitivas (de telómero a telómero). Combinando los aspectos complementarios de las tecnologías Oxford Nanopore y PacBio HiFi, 2111 nuevos genes, de los cuales 140 son codificantes, fueron descubiertos en el genoma humano cite:Nurk2021.05.26.445798.
Limitaciones de los métodos paralelos
Aunque las tecnologías de secuenciación paralelas (NGS) han revolucionado el estudio de la variedad genómica entre especies y organismos individuales, la mayoría tiene una capacidad limitada para detectar mutaciones con baja frecuencia. Este tipo de análisis es esencial para detectar mutaciones en oncogenes (genes responsables de la transformación de una célula normal a maligna), pero se ve restringido por una tasa de errores de secuenciación no despreciables, tal y como ilustra la siguiente tabla:
| Tecnología | Longitud de lectura (bp) | Tasa de error (%) |
|---|---|---|
| Sanger | ∼1,000 | 0.1–1 |
| Illumina | 2×125 | ≥0.1 |
| SOLiD | 35-50 | >0.06 |
| 454 | 400 | 1 |
| SMRT | ~10,000 | 16 |
| Ion Torrent | 400 | 1 |
Para contrarrestar este obstáculo, varias técnicas mitigatorias se han puesto en marcha. Una de las más populares es el uso de una secuencia de consenso, que es un perfil estadístico a partir de un alineamiento múltiple de secuencias. Es una forma básica de descubrimiento de patrones, en la que un alineamiento múltiple de secuencias más amplio se resume en las características que se conservan. Este tipo de análisis permite determinar la probabilidad de cada base en cada posición de una secuencia cite:10.1093/bioinformatics/btg109.

Todas las técnicas de consenso monocatenarias reducen los errores en dos o tres órdenes de magnitud, lo que es mucho mayor que cualquier enfoque computacional o bioquímico anterior, y permiten identificar con precisión variantes raras por debajo del 0.1% de abundancia. Sin embargo, persisten algunos errores. Los errores que se producen durante la primera ronda de amplificación pueden propagarse a todas las demás copias escapando la corrección cite:Salk2018.
Este problema se agrava en el análisis de repertorios inmunológicos, debido a nuestra limitada capacidad para distinguir entre la verdadera diversidad de los receptores de los linfocitos T (TCR) e inmunoglobulinas (IG) de los errores de PCR y secuenciación que son inherentes al análisis del repertorio. Las variantes resultantes de estos procesos biológicos pueden tener concentraciones drásticamente diferentes, lo que hace que aquellas con menor frecuencia sean indistinguibles de errores de secuenciación cite:Shugay2014.
Variedad genética en el sistema inmunitario
La capacidad del sistema inmunitario adaptativo para responder a cualquiera de los numerosos antígenos extraños potenciales a los que puede estar expuesta una persona depende de los receptores altamente polimórficos expresados por las células B (inmunoglobulinas) y las células T (receptores de células T [TCR]). La especificidad de las células T viene determinada principalmente por la secuencia de aminoácidos codificada en los bucles de la tercera región determinante de la complementariedad (CDR3) cite:pmid19706884.
En el timo, durante el desarrollo de los linfocitos T, se selecciona al azar un segmento génico de cada familia (V, D y J) mediante un proceso conocido como recombinación somática o recombinación V(D)J, de modo que se eliminan del genoma del linfocito los no seleccionados y los segmentos V(D)J escogidos quedan contiguos, determinando la secuencia de las subunidades del TCR y, por tanto, la especificidad de antígeno de la célula T. La selección aleatoria de segmentos junto con la introducción o pérdida de nucleótidos en sus uniones son los responsables directos de la variabilidad de TCR, cuya estimación es del orden de $10^{15}$ posibles especies distintas o clonotipos cite:BenítezCantos-Master.
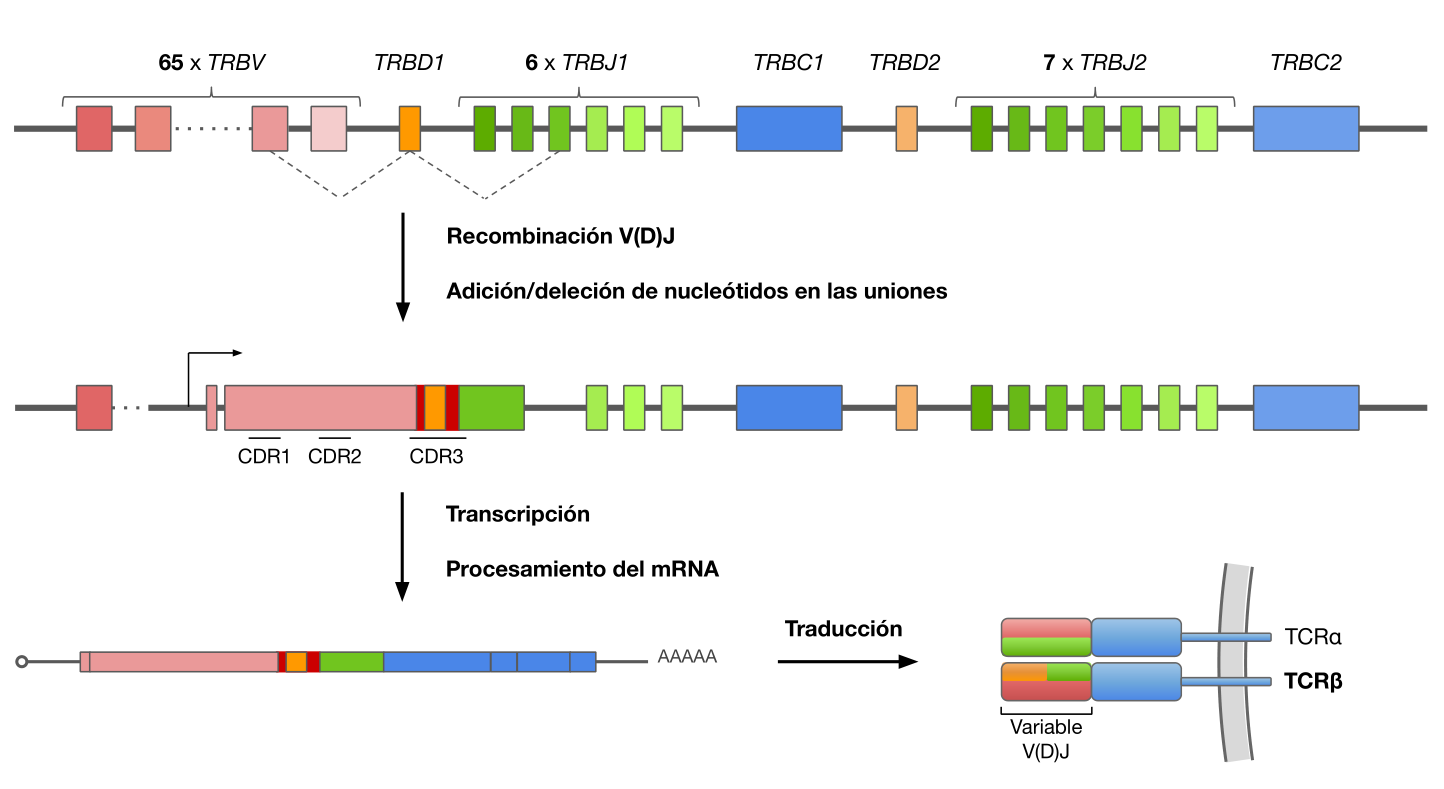
Debido a la diversidad de uniones, las moléculas de anticuerpos y TCR muestran la mayor variabilidad, formando CDR3. De hecho, debido a la diversidad de uniones, el número de secuencias de aminoácidos que están presentes en las regiones CDR3 de las de las moléculas de Ig y TCR es mucho mayor que el número que pueden ser codificadas por segmentos de genes de la línea germinal cite:abbas_lichtman_pillai_2017.
Frente a la evidencia recaudada, diversos métodos computacionales basados en la inteligencia artificial se han desarrollado para aliviar estos impedimentos.
Inteligencia artificial
La inteligencia artificial (IA) es uno de los campos más nuevos de la ciencia y la ingeniería. La investigación empezó después de la Segunda Guerra Mundial, y el término se acuñó en 1956, en la conferencia de Dartmouth College. La definición de inteligencia artificial sigue generando debate a día de hoy, por ende acotaremos la definición de inteligencia artificial al estudio de los agentes inteligentes cite📖771224.
Un agente es cualquier elemento capaz de percibir su entorno mediante sensores y actuar en consecuencia en ese entorno mediante actuadores. Un agente inteligente es aquel que actúa para conseguir el mejor resultado o, cuando hay incertidumbre, el mejor resultado esperado. En términos matemáticos, formulamos que el comportamiento de un agente se describe por la función de agente que asigna a cualquier entrada una acción cite📖771224.
Historia inicial de la inteligencia artificial
Los comienzos de la inteligencia artificial (1956-1969) se caracterizan por un entusiasmo y optimismo generalizado. Los rápidos avances en hardware, junto con el desarrollo de sistemas que redefinían las posibilidades de los ordenadores excedían las expectativas de los especialistas cite📖771224. La atmósfera frenética, durante este periodo, se puede recapitular a partir de de la propuesta de la conferencia de Dartmouth:
El estudio se basa en la conjetura de que cada aspecto del aprendizaje o cualquier otro rasgo de la inteligencia puede ser, en principio, precisamente descifrado. Se intentará encontrar un método para hacer que las máquinas utilicen el lenguaje, formen abstracciones y conceptos, resuelvan problemas que ahora están reservados a los humanos, y que se mejoren a sí mismas cite:McCarthy_Minsky_Rochester_Shannon_2006.
A finales de los años 1960, los investigadores se toparon con numerosos obstáculos al tratar de usar sus sistemas para resolver problemas más complejos. Debido a una carencia de resultados decisivos, la financiación en este ámbito académico fue cancelada cite📖771224.
Los sistemas de conocimiento (década de 1970) aprovecharon conocimiento específico de una rama para escalar los sistemas inteligentes, intercambiando sistemas generales con bajo rendimiento por sistemas específicos al problema con mejor rendimiento. Estos sistemas novedosos se empezaron a comercializar en los años 80, creando la industria de la IA cite📖771224.
Redes neuronales artificiales
Una red neuronal artificial es un modelo de computación bioinspirado, formado por capas de neuronas artificiales. Comenzaremos definiendo el concepto de neurona artificial, con el fin de introducir la noción de red neuronal artificial de forma clara y concisa.
Una neurona artificial es un modelo de una neurona biológica, cada neurona recibe un conjunto de señales y, al dispararse, transmite una señal a las neuronas interconectadas. Las entradas (inputs) se inhiben o amplifican mediante unos pesos numéricos asociados a cada conexión. El disparo, \ie activación, se controla a través de la función de activación. La neurona recoge todas las señales entrantes y calcula una señal de entrada neta en función de los pesos respectivos. La señal de entrada neta sirve de entrada a la función de activación que calcula la señal de salida cite📖80129.
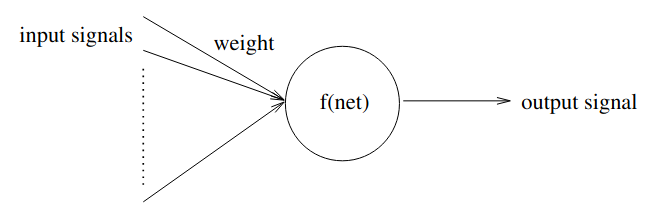
El proceso de activación se puede expresar como un modelo matemático:
\begin{equation} y= f \left(\sum\limits_{i=0}^{n} w_{i}x_{i} - T \right) \end{equation}donde $y$ es la salida del nodo, $f$ es la función de activación, $w_i$ es el peso de la entrada $x_{i}$ , y $T$ es el valor del umbral cite:Zou2009.
Una red neuronal artificial (ANN) es una red de capas de neuronas artificiales. Una ANN está formada por una capa de entrada, capas ocultas y una capa de salida. Las neuronas de una capa están conectadas, total o parcialmente, a las neuronas de la capa siguiente. También son posibles las conexiones de retroalimentación con las capas anteriores cite📖80129. La estructura típica de una ANN es la siguiente:
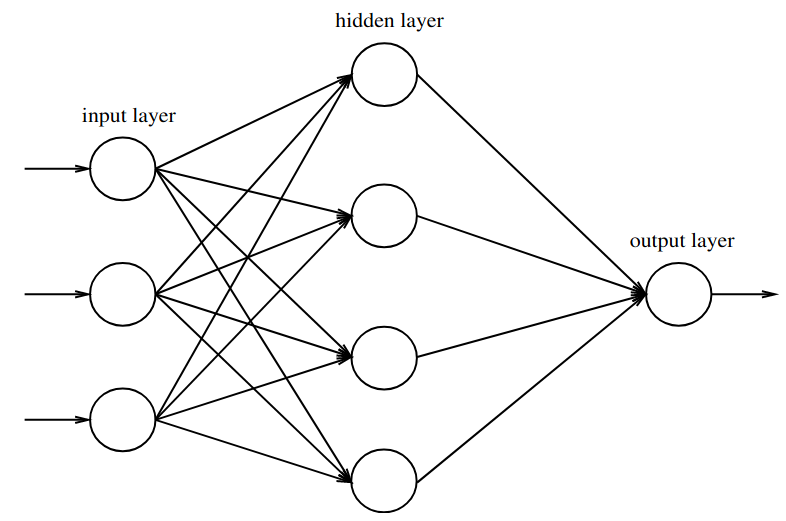
Los principios básicos de las redes neuronales artificiales fueron formulados por primera vez en 1943, y el perceptrón, que históricamente es posiblemente la primera neurona artificial, se propuso en 1958 cite📖2610592. Sin embargo, estos modelos no fueron populares hasta mediados de la década de 1980, cuando se reinventó el algoritmo de back-propagation cite📖771224.
Back propagation
El algoritmo de back propagation (BP) permite establecer los pesos y, por tanto, entrenar los perceptrones multicapa. Esto abrió el camino para el uso de las redes neuronales multicapa. La disponibilidad de un método riguroso para fijar los pesos intermedios, \ie para entrenar las capas ocultas, impulsó el desarrollo de las ANN, superando las deficiencias de la capa única propuesta por Minsky cite📖2610592. Los autores de la publicación original que describió este algoritmo sintetizan su funcionamiento a alto nivel:
Este algoritmo ajusta iterativamente los pesos de cada conexión, con el objetivo de minimizar la función de pérdida (\ie obtener la menor diferencia entre la salida de la ANN y el valor esperado). Como resultado de ello, las capas ocultas llegan a representar características importantes del dominio del problema, y las regularidades de la tarea son captadas por las interacciones de estas unidades. La capacidad de crear nuevas características útiles distingue a la retropropagación de otros métodos anteriores más sencillos, como el procedimiento de convergencia del perceptrón cite:Rumelhart1986.
Asimismo el algoritmo de back propagation es un enfoque para calcular los gradientes de forma algorítmica, que es especialmente eficaz para los modelos que emplean redes neuronales multicapa cite📖2530718. Procedemos a la exposición del algoritmo de optimización más usado con la back propagation, el gradient descent.
Gradient descent
El gradient descent es una forma de minimizar una función objetivo parametrizada por los parámetros de un modelo (e.g. pesos de una neurona artificial) mediante la actualización de los parámetros en la dirección opuesta al gradiente de la función objetivo con respecto a los parámetros, \ie seguimos la dirección de la pendiente de la superficie creada por la función objetivo cuesta abajo hasta llegar a un mínimo local o global cite:ruder2016overview.
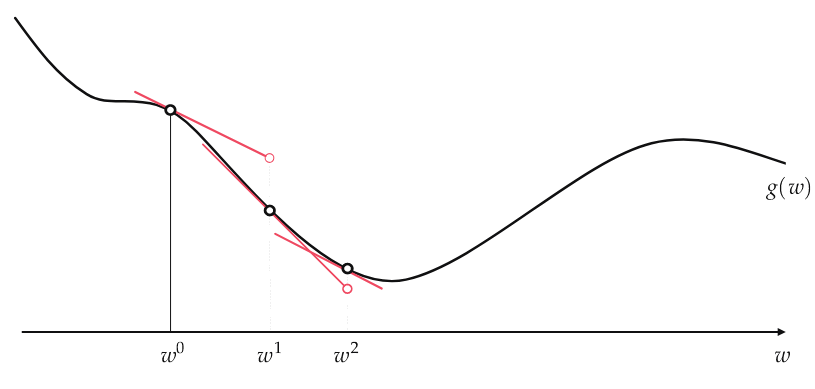
Esta secuencia de pasos se puede formular, de forma matemática:
\begin{equation} w^{k} = w^{k-1} - \alpha\nabla g(w^{k-1}) \end{equation}donde $-\nabla g(w)$ es el gradiente negativo de una función $g(w)$ en un punto cite📖2530718.
Este proceso, junto con sus variantes, es el algoritmo de optimización local más utilizado en machine learning actualmente. Esto se debe en gran medida al hecho de que la dirección de descenso proporcionada a través del gradiente es más fácil de computar (particularmente cuando la dimensión del input aumenta) cite📖2530718.
Hacia el Deep Learning
El Deep Learning es un subconjunto del machine learning, en el cual se utiliza como modelo de computación las redes neuronales artificiales (ANN), con múltiples capas ocultas. Este enfoque trata de resolver el problema de una buena capacidad expresiva de las redes neuronales, sin el aumento exponencial del número de neuronas cite:DBLP:journals/corr/WangRX17.
La pregunta sobre cómo mantener esta capacidad expresiva de la red y al mismo tiempo reducir el número de unidades de cálculo se ha planteado durante años. Intuitivamente, se sugirió que es natural buscar redes más profundas porque 1) el sistema neuronal humano es una arquitectura profunda y 2) los humanos tienden a representar conceptos en un nivel de abstracción como la composición de conceptos en niveles inferiores. Actualmente, la solución es construir arquitecturas más profundas, \ie redes neuronales con más capas ocultas cite:DBLP:journals/corr/WangRX17.
En la actualidad, los avances tanto en potencia de cálculo del hardware, especialmente en las tarjetas gráficas (GPU) cite:Cireşan2010, junto con la disponibilidad de grandes datasets cite📖771224 han permitido que esta disciplina prospere.
Este área de conocimiento ha mejorado drásticamente el estado del arte en muchas tareas de inteligencia artificial, como la detección de objetos, el reconocimiento del habla o la traducción automática. Su naturaleza de arquitectura profunda otorga al aprendizaje profundo la posibilidad de resolver muchas tareas de IA más complicadas cite:DBLP:journals/corr/WangRX17. Como resultado, los investigadores están extendiendo el aprendizaje profundo a una variedad de diferentes dominios y tareas modernas:
- Eliminación del ruido en las señales de voz
- Descubrimiento de patrones de agrupación de expresiones genéticas
- Generación de imágenes con distintos estilos
- Análisis de sentimientos de diferentes fuentes simultáneas
Bioinformática
La bioinformática es un campo interdisciplinar en el que intervienen principalmente la biología molecular y la genética, la informática, las matemáticas y la estadística. Los problemas biológicos a gran escala y con gran cantidad de datos se abordan desde un punto de vista informático. Los problemas más comunes son el modelado de procesos biológicos a nivel molecular y la realización de inferencias a partir de los datos recogidos, cite:Can2014 en particular:
| Problema | Fuente de datos |
|---|---|
| Alineamiento de secuencias múltiples | ADN |
| Búsqueda de patrones de secuencias | ADN |
| Análisis evolutivos | ADN |
| Análisis de la función | Proteínas |
| Predicción de las estructuras | Proteínas |
| Estudio de las vías metabólicas | Redes biológicas |
Es tentador atribuir los orígenes de la bioinformática a la reciente convergencia de la secuenciación del ADN, los proyectos genómicos a gran escala, Internet y los superordenadores. Sin embargo, algunos científicos que afirman que la bioinformática se encuentra en su infancia reconocen que los ordenadores eran herramientas importantes en la biología molecular una década antes de que la secuenciación del ADN se convirtiera en algo factible (década de los 1960) cite:Hagen2000.
Estado del arte
Procedemos a realizar un estudio de las metodologías actuales en los ámbitos, introducidos previamente, del Deep Learning y de la bioinformática, con el objetivo de identificar las técnicas que se utilizan a nivel académico y en la industria.
Deep Learning
El gran potencial de las ANN es la alta velocidad de procesamiento que ofrecen en una implementación paralela masiva, lo que ha aumentado la necesidad de investigar en este ámbito. Hoy en día, las ANN se utilizan sobre todo para la aproximación de funciones universales en paradigmas numéricos debido a sus excelentes propiedades de autoaprendizaje, adaptabilidad, tolerancia a los fallos, no linealidad y avance en el mapeo de la entrada a la salida. Las ANN son capaces de resolver aquellos problemas que no pueden resolverse con la capacidad de cómputo de los procedimientos tradicionales y las matemáticas convencionales cite:ABIODUN2018e00938.
Los métodos de Deep Learning han resultado ser adecuados para el estudio de big data con un éxito notable en su aplicación al reconocimiento del habla, computer vision, el reconocimiento de patrones, los sistemas de recomendación y el procesamiento del lenguaje natural (NLP) cite:LIU201711. En la actualidad, la innovación de DL en la identificación de imágenes, la detección de objetos, la clasificación de imágenes y las tareas de identificación de rostros tienen un gran éxito cite:ABIODUN2018e00938.
En nuestro estudio, evaluaremos 2 arquitecturas de Deep Learning: autoencoder y CNN. Estableceremos una comparación entre estas diferentes estructuras de ANN, además de mencionar avances recientes en estos algoritmos.
Autoencoder
Un autoencoder (AE) es un tipo de ANN, se trata de un algoritmo de aprendizaje no supervisado que se utiliza para codificar eficazmente un conjunto de datos con el fin de reducir la dimensionalidad. Durante las últimas décadas, los AE han estado a la vanguardia en el ámbito del Deep Learning. Los datos de entrada se convierten primero en una representación abstracta que, a continuación, la función codificadora vuelve a convertir en el formato original. En concreto, se entrena para codificar la entrada en alguna representación de modo que la entrada pueda reconstruirse a partir de esa representación cite:LIU201711
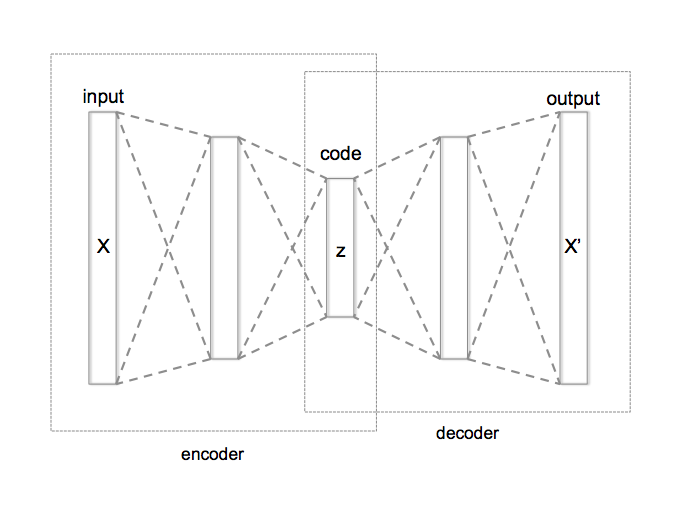
Un autoencoder se compone de 2 elementos:
| Elemento | Función |
|---|---|
| Encoder | $h = f(x)$ |
| Decoder | $r = g(h)$ |
El proceso de aprendizaje se describe como una optimización de una función de pérdida:
\begin{equation} L(x, g(f(x))) \end{equation}Donde $L$ es la función de pérdida que penaliza $g(f(x))$ por ser distinto de $x$ cite:Goodfellow-et-al-2016.
Tradicionalmente, los autoencoders se utilizaban para reducir la dimensionalidad o feature learning. Recientemente, ciertas teorías que conectan los AE y los modelos de variables latentes han llevado a los autocodificadores a la vanguardia del modelado generativo cite:Goodfellow-et-al-2016.
En la actualidad, los autoencoders se utilizan para la reducción de ruido, tanto en texto cite:Lewis_2020 como en imágenes cite:bigdeli17_image_restor_using_autoen_prior, clustering no supervisado cite:makhzani15_adver_autoen, generación de imágenes sintéticas cite:Yoo_2020, reducción de dimensionalidad cite:makhzani15_adver_autoen, predicción de secuencia a secuencia para la traducción automática cite:kaiser18_discr_autoen_sequen_model.
Objetivos
- Introducción al dominio de un problema de biología molecular: Secuenciación de ADN y análisis de receptores de linfocitos T (TCR)
- Introducción al análisis bioinformático de secuencias de ADN: preprocesamiento de lecturas, alineamiento y otros análisis bioinformáticos asociados.
- Creación de un repositorio software para la generación in-silico de secuencias de TCR y la simulación de la secuenciación de las mismas.
- Introducción al uso de Tensorflow y Keras para Deep Learning
- Estudio de aplicación de Tensorflow/Keras a la corrección de errores de secuenciación en base a los datos sintetizados previamente.