Compare commits
2 Commits
e35f8a6dee
...
abed2c2666
| Author | SHA1 | Date | |
|---|---|---|---|
|
abed2c2666
|
|||
|
5f9951ec19
|
@ -29,17 +29,47 @@ Next generation sequencing (NGS) have revolutionised genomic research. These tec
|
||||
|
||||
* Introducción
|
||||
|
||||
** Secuenciación de ADN
|
||||
|
||||
La secuenciación de ADN es el proceso mediante el cual se determina el orden de los nucleótidos en una secuencia de ADN. En los años 70, Sanger \etal desarrollaron métodos para secuenciar el ADN mediante técnicas de terminación de cadena. cite:Sanger5463 Este avance revolucionó la biología, proporcionando las herramientas necesarias para descifrar genes, y posteriormente, genomas completos. La demanda creciente de un mayor rendimiento llevó a la automatización y paralelización de las tareas de secuenciación. Gracias a estos avances, la técnica de Sanger permitió determinar la primera secuencia del genoma humano en 2004 (Proyecto Genoma Humano). cite:InternationalHumanGenomeSequencingConsortium2004
|
||||
|
||||
Sin embargo, el Proyecto Genoma Gumano requerió una gran cantidad de tiempo y recursos, y era evidente que se necesitaban tecnologías más rápidas, de mayor rendimiento y más baratas. Por esta razón, en el mismo año (2004) el /National Human Genome Research Institute/ (NHGRI) puso en marcha un programa de financiación con el objetivo de reducir el coste de la secuenciación del genoma humano a 1000 dólares en diez años. cite:Schloss2008 Esto estimuló el desarrollo y la comercialización de las tecnologías de secuenciación de alto rendimiento o /Next-Generation Sequencing/ (NGS), en contraposición con el método automatizado de Sanger, que se considera una tecnología de primera generación.
|
||||
|
||||
** Técnicas de secuenciación de alto rendimiento
|
||||
|
||||
Estos nuevos métodos de secuenciación proporcionan tres mejoras importantes: en primer lugar, en lugar de requerir la clonación bacteriana de los fragmentos de ADN, se basan en la preparación de bibliotecas de moléculas en un sistema sin células. En segundo lugar, en lugar de cientos, se producen en paralelo de miles a muchos millones de reacciones de secuenciación. Finalmente, estos resultados de secuenciación se detectan directamente sin necesidad de electroforesis. cite:vanDijk2014
|
||||
|
||||
\clearpage
|
||||
|
||||
Actualmente, se encuentran en desarrollo las tecnologías de tercera generación de secuenciación (Third-Generation Sequencing). Existe un debate considerable sobre la diferencia entre la segunda y tercera generación de secuenciación, la secuenciación en tiempo real y la divergencia simple con respecto a las tecnologías anteriores deberían ser las características definitorias de la tercera generación. Aquí consideramos que las tecnologías de tercera generación son aquellas capaces de secuenciar moléculas individuales, negando el requisito de amplificación del ADN que comparten todas las tecnologías anteriores. cite:HEATHER20161
|
||||
|
||||
# Not in the correct place
|
||||
Estas nuevas técnicas han demostrado su valor, con avances que han permitido secuenciar el genoma humano completo, incluyendo las secuencias repetitivas (de telómero a telómero). Combinando los aspectos complementarios de las tecnologías Oxford Nanopore y PacBio HiFi, 2111 nuevos genes, de los cuales 140 son codificantes, fueron descubiertos en el genoma humano. cite:Nurk2021.05.26.445798
|
||||
|
||||
** Limitaciones de los métodos paralelos
|
||||
|
||||
Aunque las tecnologías de secuenciación paralelas (NGS) han revolucionado el estudio de la variedad genómica entre especies y organismos individuales, la mayoría tiene una capacidad limitada para detectar mutaciones con baja frecuencia. Este tipo de análisis es esencial para detectar mutaciones en oncogenes (genes responsables de la transformación de una célula normal a maligna), pero se ve restringido por una tasa de errores de secuenciación no despreciables. En 2011, la tasa de errores por substitución (intercambio de un nucleótido por otro) era > 0.1%, y era similar en estudios posteriores. cite:Ma2019
|
||||
|
||||
#+CAPTION: Tasa de error de secuenciación global de dos secuenciadores NovaSeq en la biblioteca de ADN de referencia COLO829. La mediana de las tasas de error (barras negras verticales) de cada tipo de desincorporación en bases de tipo salvaje conocidas se indica en el margen izquierdo del panel. cite:Davis2021
|
||||
#+ATTR_HTML: :height 30% :width 50%
|
||||
#+NAME: fig:sequencing-errors
|
||||
[[./assets/figures/sequencing-errors.png]]
|
||||
|
||||
Este problema se agrava en el análisis de repertorios inmunológicos, debido a nuestra limitada capacidad para distinguir entre la verdadera diversidad de los receptores de los linfocitos T (TCR) e inmunoglobulinas (IG) de los errores de PCR y secuenciación que son inherentes al análisis del repertorio. Los clonotipos resultantes pueden tener concentraciones drásticamente diferentes, lo que hace que los clonotipos menores sean indistinguibles de las variantes erróneas. cite:Shugay2014
|
||||
|
||||
** Variedad genética en el sistema inmunitario
|
||||
|
||||
La capacidad del sistema inmunitario adaptativo para responder a cualquiera de los numerosos antígenos extraños potenciales a los que puede estar expuesta una persona depende de los receptores altamente polimórficos expresados por las células B (inmunoglobulinas) y las células T (receptores de células T [TCR]). La especificidad de las células T viene determinada principalmente por la secuencia de aminoácidos codificada en los bucles de la tercera región determinante de la complementariedad (CDR3). cite:pmid19706884
|
||||
|
||||
En el timo, durante el desarrollo de los linfocitos T, se selecciona al azar un segmento de cada familia mediante un proceso conocido como recombinación somática o recombinación V(D)J, de modo que se eliminan del genoma del linfocito los no seleccionados y los segmentos V(D)J escogidos quedan contiguos, determinando la secuencia de las subunidades del TCR y, por tanto, la especificidad de antígeno de la célula T. La selección aleatoria de segmentos junto con la introducción o pérdida de nucleótidos en sus uniones son los responsables directos de la variabilidad de TCR, cuya estimación es del orden de $10^{15}$ posibles especies distintas o clonotipos. cite:BenítezCantos-Master
|
||||
|
||||
#+CAPTION: Generación de diversidad en el TCR \alpha \beta. Durante el desarrollo de los linfocitos T se reordenan los segmentos génicos V (rosa), D (naranja) y J (verde) a través del proceso de recombinación V(D)J. Durante este proceso se pueden añadir o eliminar nucleótidos en la unión de los segmentos (rojo), contribuyendo a la diversidad de la secuencia. Se señalan las 3 regiones CDR, estando CDR3 localizada en la unión V(D)J. cite:BenítezCantos-Master
|
||||
#+ATTR_HTML: :height 30% :width 90%
|
||||
#+NAME: fig:vdj-recombination
|
||||
[[./assets/figures/VDJ.png]]
|
||||
|
||||
Debido a la diversidad de uniones, las moléculas de anticuerpos y TCR muestran la mayor variabilidad, que forman la región determinante de la complementariedad 3 (CDR3). De hecho, debido a la diversidad de uniones, el número de secuencias de aminoácidos que están presentes en las regiones CDR3 de las de las moléculas de Ig y TCR es mucho mayor que el número que pueden ser codificadas por segmentos de genes de la línea germinal. cite:abbas_lichtman_pillai_2017
|
||||
|
||||
|
||||
* Estado del arte
|
||||
** Bioinformática
|
||||
** Deep Learning
|
||||
|
||||
BIN
Dissertation.pdf
BIN
Dissertation.pdf
Binary file not shown.
@ -463,6 +463,8 @@
|
||||
|
||||
\frontmatter
|
||||
\tableofcontents
|
||||
\listoftables{}
|
||||
\listoffigures{}
|
||||
\mainmatter{}
|
||||
$body$
|
||||
\backmatter{}
|
||||
|
||||
@ -342,3 +342,75 @@
|
||||
doi = {10.1038/nbt1008-1113},
|
||||
url = {https://doi.org/10.1038/nbt1008-1113}
|
||||
}
|
||||
|
||||
@Article{Shugay2014,
|
||||
author = {Shugay, Mikhail and Britanova, Olga V. and Merzlyak,
|
||||
Ekaterina M. and Turchaninova, Maria A. and Mamedov, Ilgar Z.
|
||||
and Tuganbaev, Timur R. and Bolotin, Dmitriy A. and
|
||||
Staroverov, Dmitry B. and Putintseva, Ekaterina V. and
|
||||
Plevova, Karla and Linnemann, Carsten and Shagin, Dmitriy and
|
||||
Pospisilova, Sarka and Lukyanov, Sergey and Schumacher, Ton N.
|
||||
and Chudakov, Dmitriy M.},
|
||||
title = {Towards error-free profiling of immune repertoires},
|
||||
journal = {Nature Methods},
|
||||
year = 2014,
|
||||
month = {Jun},
|
||||
day = 01,
|
||||
volume = 11,
|
||||
number = 6,
|
||||
pages = {653-655},
|
||||
abstract = {A two-step error correction process for high
|
||||
throughput--sequenced T- and B-cell receptors allows the
|
||||
elimination of most errors while not diminishing the natural
|
||||
complexity of the repertoires.},
|
||||
issn = {1548-7105},
|
||||
doi = {10.1038/nmeth.2960},
|
||||
url = {https://doi.org/10.1038/nmeth.2960}
|
||||
}
|
||||
|
||||
@Article{Ma2019,
|
||||
author = {Ma, Xiaotu and Shao, Ying and Tian, Liqing and Flasch,
|
||||
Diane A. and Mulder, Heather L. and Edmonson, Michael N. and
|
||||
Liu, Yu and Chen, Xiang and Newman, Scott and Nakitandwe, Joy
|
||||
and Li, Yongjin and Li, Benshang and Shen, Shuhong and Wang,
|
||||
Zhaoming and Shurtleff, Sheila and Robison, Leslie L. and
|
||||
Levy, Shawn and Easton, John and Zhang, Jinghui},
|
||||
title = {Analysis of error profiles in deep next-generation
|
||||
sequencing data},
|
||||
journal = {Genome Biology},
|
||||
year = 2019,
|
||||
month = {Mar},
|
||||
day = 14,
|
||||
volume = 20,
|
||||
number = 1,
|
||||
pages = 50,
|
||||
abstract = {Sequencing errors are key confounding factors for detecting
|
||||
low-frequency genetic variants that are important for cancer
|
||||
molecular diagnosis, treatment, and surveillance using deep
|
||||
next-generation sequencing (NGS). However, there is a lack of
|
||||
comprehensive understanding of errors introduced at various
|
||||
steps of a conventional NGS workflow, such as sample handling,
|
||||
library preparation, PCR enrichment, and sequencing. In this
|
||||
study, we use current NGS technology to systematically
|
||||
investigate these questions.},
|
||||
issn = {1474-760X},
|
||||
doi = {10.1186/s13059-019-1659-6},
|
||||
}
|
||||
|
||||
@mastersthesis{BenítezCantos-Master,
|
||||
author = "María Soledad Benítez Cantos",
|
||||
title = "Análisis de repertorios de receptores de células T a partir de datos de secuenciación masiva",
|
||||
school = "Universidad de Granada",
|
||||
year = "2019",
|
||||
month = "{Jul}",
|
||||
}
|
||||
|
||||
@inbook{abbas_lichtman_pillai_2017,
|
||||
place = {Philadelphia, PA},
|
||||
edition = {9th},
|
||||
booktitle = {Cellular and molecular immunology},
|
||||
publisher = {Elsevier},
|
||||
author = {Abbas, Abul K. and Lichtman, Andrew H. and Pillai, Shiv},
|
||||
year = 2017,
|
||||
pages = 204
|
||||
}
|
||||
|
||||
BIN
assets/figures/VDJ.png
Normal file
BIN
assets/figures/VDJ.png
Normal file
{kind=link}
Binary file not shown.
|
After 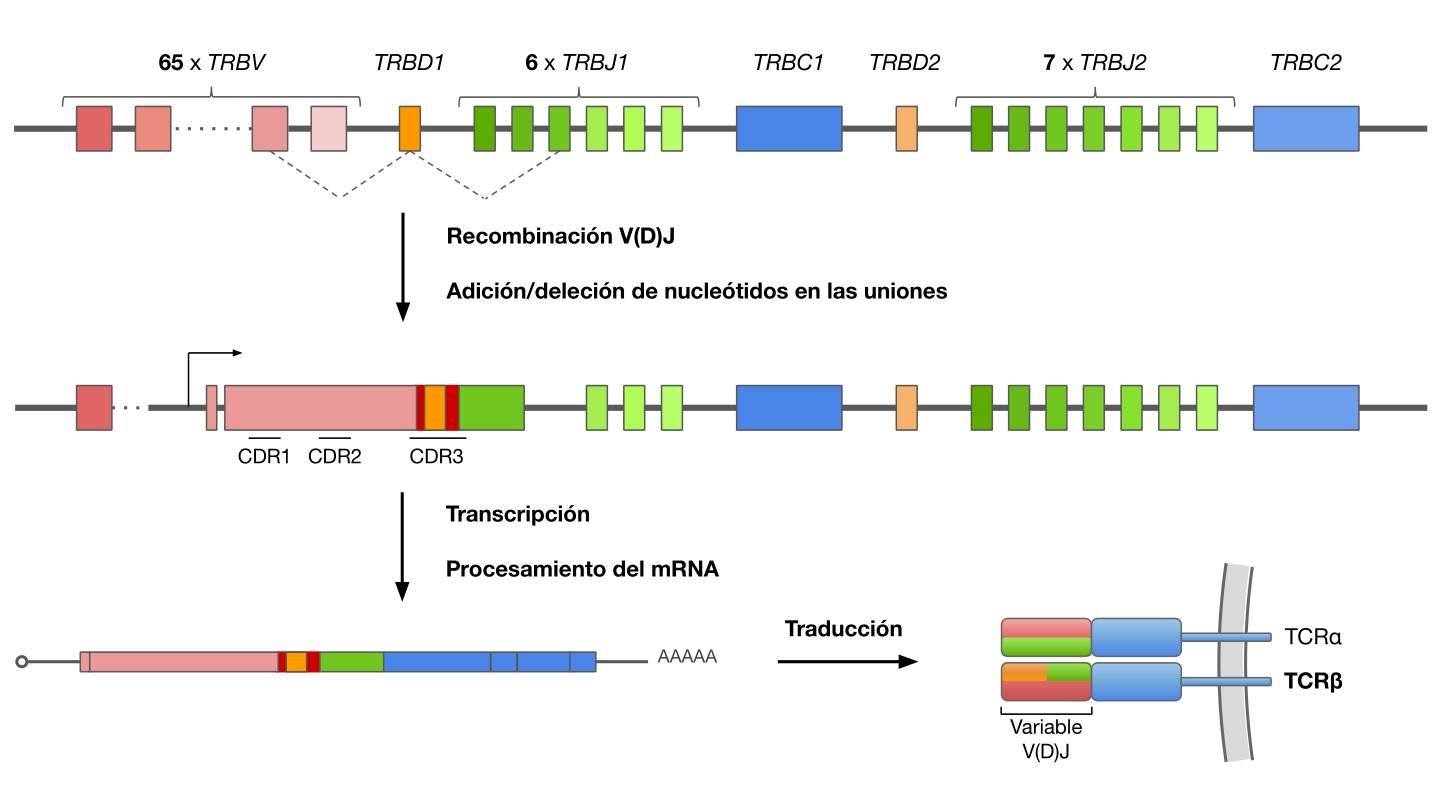
(image error) Size: 64 KiB |
BIN
assets/figures/sequencing-errors.png
Normal file
BIN
assets/figures/sequencing-errors.png
Normal file
{kind=link}
Binary file not shown.
|
After 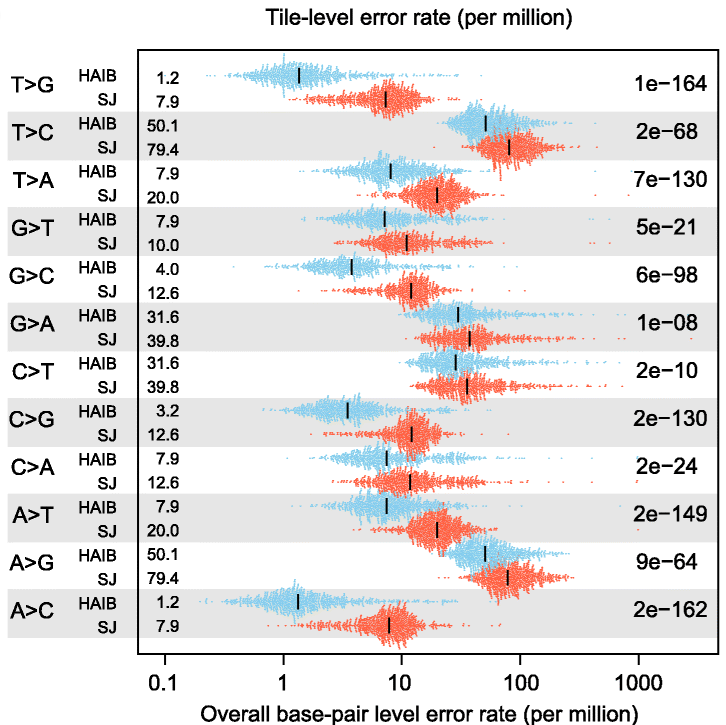
(image error) Size: 162 KiB |
Loading…
Reference in New Issue
Block a user